

Many companies developing healthcare software don't realize they qualify as SaMD manufacturers. Unlike its traditional hardware counterparts, software as a medical device exists entirely as software, yet its patient safety risk profile is considered to mirror that of traditional hardware-based medical devices. This duality creates a unique regulatory precedent, complicating matters for companies that want to place SaMD products on global markets.
If you’re also feeling overwhelmed by a patchy regulatory landscape, our health tech team has created this guide to clarify the regulatory and classification complexities of SaMD.
What is SaMD (Software as a Medical Device)?
As the International Medical Device Regulators Forum (IMDRF) puts it, software as a medical device is software designed to be used for medical reasons all on its own, not as a built-in part of a physical medical device.
Yet, there is still no definitive answer to the question of what SaMD is. Each country has its own regulatory body, with many having a different understanding of what SaMD is.
According to the IMDFR guidelines, the SaMD category also includes:
- Software-based in vitro devices.
- Software that can operate beyond medical-purpose computing platforms.
- Software that’s capable of working in tandem with other products, including other medical devices, general-purpose software, and other SaMD software.
- Mobile apps that qualify as SaMD.
However, software that controls or operates a hardware medical device is not considered SaMD according to the IMDRF.
According to the IMDRF definition, an AI mobile app that analyzes user-submitted photos of skin lesions to estimate the risk rate of melanoma is considered SaMD. But an AI mobile app that logs daily water intake is not SaMD as it’s not intended for diagnosis or treatment (i.e., lacks the intended medical purpose of the SaMD).
Other examples of SaMD include:
- AI-enabled software that detects cardiac arrhythmias based on ECG data.
- IVD software that identifies genetic markers for diseases based on the analysis of genomic data.
- Remote patient monitoring applications that track vital signs and notify care teams of potential problems.
- Mental health apps that offer personalized interventions based on the analysis of mood, anxiety, or depression symptoms.
Note that the IMDRF is a voluntary group of global medical device regulators, so these guidelines do not have legal authority in all jurisdictions. However, as the International Medical Device Regulators Forum is chaired by the FDA, its recommendations still carry significant weight worldwide, especially in regions with developing regulatory frameworks.
What’s the difference between SaMD and SiMD? Examples included
Software as a Medical Device is just one of the three types of software related to medical devices. The FDA also singles out the other two types of software that are directly or indirectly related to medical devices. These are Software in a medical device (SiMD) and software used by medical device manufacturers for production and upkeep.
As upkeep and production software is outside the diagnosis, treatment, and disease prevention field, its regulatory pathway doesn’t overlap with SaMD. Conversely, both SaMD and SiMD are designed for medical purposes, which complicates their classification in complex interactions.
Our team has prepared a comparison table to highlight the main differences and make it easier for you to differentiate between SaMD and SiMD.
| Factor | Software as a medical device (SaMD) | Software in a medical device (SiMD) |
|---|---|---|
| Independence from hardware | Runs independently on a general-purpose computing platform. | Is either built into a hardware medical device or relies on it to fulfill its function. |
| Main function | Can diagnose, offer personalized treatments, and inform clinical management, i.e., performs medical functions directly. | Used for controlling the medical functions of the hardware device. |
| Regulatory status | Regulated as a standalone medical device. | Regulated as a component of the hardware medical device. |
| Update mechanism | Software updates can be deployed independently. | Updates can necessitate changes to the hardware device or re-certification. |
| Examples | • Insulin dosing calculators • AI disease risk prediction tools • AI medical image analysis applications • Cardiac arrhythmia detection app | • Software within implantable medical devices (pacemakers, neurostimulator) • Software within infusion pumps • Software within imaging devices • Software within monitoring devices (glucose meters, wearable EKG monitors, imaging devices) |
According to the FDA, the regulatory requirements for SaMD are based on its independent function and data analysis. SiMD, on the other hand, is regulated as a component within the larger medical device, with its safety and effectiveness being evaluated through the lens of the overall device's performance.
Key regulatory frameworks for SaMD by region
The global regulatory area for SaMD is divided into distinct key regions, each with a specific set of regulatory standards and frameworks. A common thread between all SaMD regulations is their risk-based approach to SaMD classification. However, each regulation has a different interpretation of what risk is and how much scrutiny different risk levels are subject to.
SaMD within the IMDRF guidelines (international)
The IMDRF developed its regulatory framework for SaMD to provide harmonized, overarching guidelines for individual jurisdictions to tailor to their own regulatory structure. Although the IMDRF guidelines are non-mandatory, many regional regulatory bodies, including the FDA in the US and the MDR in the EU, use them as a point of reference.
Previously, the IMDRF classification for SaMD revolved around the ‘levels of concern’ concept that grouped SaMD into minor, moderate, and major risk categories, based on potential patient harm.
Today, the guidelines are driven by a different classification approach that assigns SaMD one of the four categories (I, II, III, IV) depending on the SaMD’s impact on the patient or public health in specific healthcare scenarios.
| State of healthcare situation or condition | Significance of information provided by SaMD to the healthcare decision | ||
|---|---|---|---|
| Treat or diagnose | Drive clinical management | Inform clinical management | |
| Critical | IV | III | II |
| Serious | III | II | I |
| Non-serious | II | I | I |
Category IV has the highest level of impact (hence, the highest risk level), while Category I has the lowest impact. In other words, the more critical the situation and the more directly the SaMD impacts patient outcomes, the higher the risk category and the stricter the regulatory scrutiny is.
Criteria for Category I
The lowest-risk category includes software that informs clinical management in a serious or non-serious situation, as opposed to dictating the course of action. Because it doesn’t have a direct impact on the patient’s well-being, Category I SaMD is considered to be of low impact.
Example: a mobile app for medication tracking that doesn't analyze the data to provide treatment advice or a SaMD that collates historical blood pressure data for a clinician’s review.
Criteria for Category II
Classified as medium impact, Category II SaMD operates to treat or diagnose a disease in a non-serious situation or to drive clinical disease management in a serious situation. In critical situations, Category II SaMD informs clinical management for a disease or condition.
Example: a medication mobile app for chronic disease patients that analyzes medication adherence data, sends reminders, and identifies potential missed doses.
Criteria for Category III
A SaMD is considered to fall into this category if it offers vital information for treating, diagnosing, or managing a disease in a serious situation, and is considered to have a high impact on the patient’s health.
Example: SaMD that analyzes CGM data and automatically modifies the insulin pump settings within predefined safety parameters to keep blood sugar under control in patients with type 1 diabetes.
Criteria for Category IV
Any SaMD that informs disease/condition treatment or diagnosis in a critical situation and is considered to be of very high impact belongs to the fourth category.
Example: SaMD that keeps tabs on a patient’s vital signs in ICUs, automatically triggers immediate alerts, and recommends specific interventions based on detected critical thresholds that, if acted upon incorrectly or delayed, could lead to immediate death or irreversible organ damage.
SaMD within the FDA regulatory framework (the US)
The FDA’s take on SaMD is largely impacted by the IMDRF guidelines. Both agencies share the definition of SaMD, the risk-based categorization principle, and guidance on clinical evaluation.
What’s different, however, is that the FDA has its own three-class SaMD classification system (Class I, Class II, Class III) based on the intended use of the device and the risk to the patient if the device malfunctions or provides inaccurate information. The FDA’s SaMD classification aligns with the classification for traditional medical devices.
The device’s classification also defines the documentation level SaMD manufacturers require for a premarket submission: basic or enhanced. Also, keep in mind that the FDA classification system is predicate-based, meaning that the FDA classifies new devices based on similar devices that are already on the U.S. market.

Class I: Low to medium risk
Class I SaMD has the lowest potential for harmfully affecting the patient’s or user’s health. These are usually simple in design and focused on collecting data without making the data actionable. From a regulatory standpoint, class I SaMD is exempt from Premarket Notification (510(k)), relieving manufacturers of the obligation to submit data to the FDA for clearance before marketing their device.
Class I SaMD is also subject to general controls that include device listing, establishment registration, implementation of quality systems, and other basic regulatory requirements. According to the FDA, 47% of medical devices belong to this category, with 95% of these being outside the scope of the regulatory process.
Example: SaMD that collects manually entered historical blood pressure data and visualizes it for the user’s personal review, with no treatment recommendations or independent interpretation.
Class II: Medium to high risk
Class II devices have a more far-reaching effect on someone’s health if they develop a fault. As medium- or high-risk devices, class II SaMD are bound by general controls plus special controls, which means that they may require additional software-related documentation in a premarket submission. Plus, most class II devices require Premarket Notification (510(k) Clearance).
Example: SaMD that analyzes sleep patterns from wearable sensors, delivers a snapshot of the user’s sleep quality and stages, and suggests lifestyle changes without formulating a diagnosis.
📌 If your SaMD is novel in design or intended use and there are no legally marketed predicate devices to associate it with, you can submit a De Novo request for the FDA to classify your device into class I or II.
Class III: High risk
To be classified as class III, SaMD must be associated with a high risk to the patient or the user. Devices in this category potentially have far-reaching consequences if they fail to function as intended or provide misleading information. Class III SaMD is subject to general controls and the Premarket Approval (PMA) process, where manufacturers must validate the safety and effectiveness of their devices with valid scientific evidence collected from human clinical trials.
Example: SaMD that analyzes genomic data, identifies a patient’s immediate risk of developing an aggressive cancer, and recommends high-risk treatment strategies.
📌 If you are unsure which class your SaMD belongs to, you can ask the FDA for a formal classification process through the 513(g) Request. However, this option has a fee and can take a while.
SaMD through the prism of the MDR and IVDR frameworks (the EU)
If you're planning on placing your SaMD on the EU market, you will need to comply with the requirements of the EU Medical Device Regulation (MDR) or the EU In Vitro Diagnostic Medical Devices Regulation (IVDR) based on the intended purpose of your software product.
The EU doesn’t apply the term “SaMD” and uses the term “medical device software” or “MDSW” instead. The software is filed under the definition of a medical device or an in vitro diagnostic medical device if it directly influences medical decisions or patient outcomes.
The EU Medical Device Regulation
Within the MDR, Rule 11 specifically addresses the classification of software that functions as a medical device, i.e., used for diagnosis, treatment decisions, or physiological monitoring. According to Rule 11, MDSWs are mapped to the classes I, IIa, IIb, or III based on the severity of potential harm.
Summing up Rule 11's key points:
- Software used to make diagnosis/treatment decisions is generally classified as class IIa.
- If those diagnosis/treatment decisions can potentially result in death or irreversible health damage, it's class III.
- Software is associated with class IIb if those decisions could be the reason behind serious health damage or surgery.
- Software that monitors physiological processes is generally class IIa.
- However, if it monitors vital signs where changes could cause immediate danger, it's class IIb.
- All other software is class I (lowest risk).
The flip side of this classification is that an MDSW with an even low actual risk could be classified as Class III just because it can lead to irreversible damage in a very unlikely worst-case scenario.
📌 To clarify the application of Rule 11 and the risk categorization, the Medical Device Coordination Group (MDCG) has issued several guidance documents. While not legally binding, the MDCG guidelines provide a harmonized understanding of how EU authorities interpret MDSW and how they expect the MDR to be implemented.
The EU In Vitro Diagnostic Medical Devices Regulation (IVDR)
Your MDSW is subject to the IVDR if it’s designed to gather data from tests performed on human samples (like blood, urine, tissues) and analyze this data to suggest interpretations about those samples for a medical purpose.
At the core of the IVDR classification system is the potential risk of an in vitro diagnostic device to public health and individuals. It divides MDSWs into four classes:
- Class A (low individual and public health risk) has no direct impact on test results.
- Class B (moderate individual and/or low public health risk) includes software for self-testing, blood grouping, and certain infectious disease tests.
- Class C (high individual and/or moderate public health risk) includes software for cancer screening, genetic testing, and high-impact infectious disease tests.
- Class D (high individual and high public health risk) includes software for detecting high-risk transmissible agents and for critical blood group compatibility testing.
📌 Like in the MDR, the MDCG acts as the central coordinating body for the practical implementation of the IVDR across the EU, providing the necessary guidelines.
Software safety classification of IEC 62304 (international)
IEC 62304 is an international standard for medical device development issued by the International Electrotechnical Commission (IEC). Concerned with building safe and effective software, IEC 62304 details life cycle requirements for developing medical software and software within medical devices.
On a higher level, IEC 62304 establishes a common way of thinking for the global medical device development community. Both the FDA and the EU recognize this standard and often consider compliance with IEC 62304 as a strong indicator of adherence to their guidelines.
The IEC 62304 classification system is based on the likelihood of the SaMD to contribute to hazardous situations and includes three software safety levels:
- Class A — SaMD whose failure could not possibly lead to injury or damage to health.
- Class B — SaMD whose failure could possibly lead to non-serious injury.
- Class C — SaMD whose failure could possibly lead to death or serious injury.
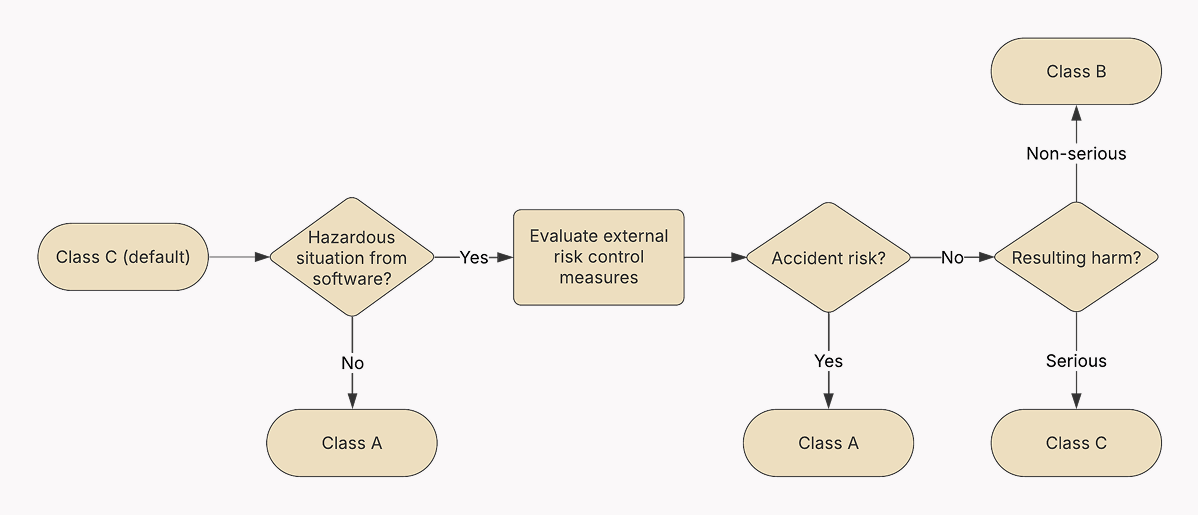
📌 The IEC 62304 standard does not equate to the SaMD safety. Instead, it helps you identify the right level of rigor for your development process. The higher the potential severity of harm associated with a SaMD failure, the more detailed, documented, and validated the entire software development lifecycle must be according to IEC 62304.
While you can’t directly map the IEC 62304 software safety classes to the FDA’s or the MDR’s classification, there can be some overlap between them. For example, higher-risk SaMD (FDA Class III, EU MDR Class III) are more likely to contain software classified as IEC 62304 Class C. At the same time, lower-risk SaMD (FDA Class I, EU MDR Class I) are more likely to contain software classified as IEC 62304 Class A or B.
Still, there is no direct correspondence. That’s why we recommend viewing IEC 62304 compliance as a way to mitigate software-related risks within SaMD, regardless of the overall device classification.
SaMD software development process according to IEC 62304
As most regulatory bodies recognize IEC 62304 as a consensus process standard and view IEC 62304-compliant development process as a sign of meeting their requirements, it makes sense to organize your SDLC around it.
📌 Although IEC 62304 is structured in a sequential manner, your SDLC doesn’t have to follow the Waterfall model to be compliant with it. Any approach, including Agile, is fine as long as you don’t skip the activities and requirements listed in IEC 62304.
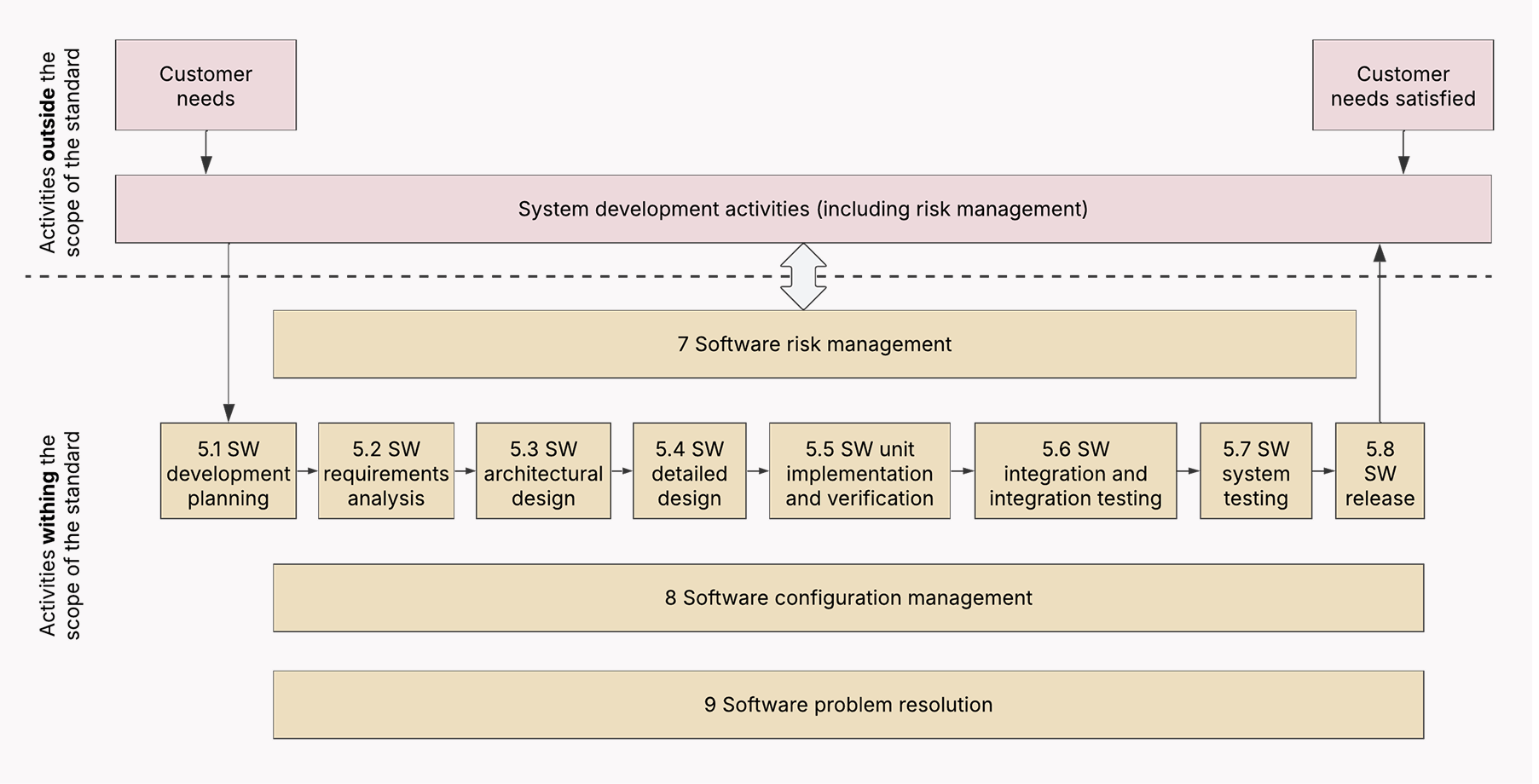
Below, we’ve listed the software development steps you need to include in your development process to comply with the IEC 62304 standard.
Phase 1: Planning
The IEC 62304 standard assumes that your medical device software is built and maintained on the solid foundation of a quality management system (QMS) and a risk management system. So the planning stage focuses on laying the groundwork for compliant development.
- Set up a Quality Management System (QMS) — Make sure your development team establishes a QMS with processes mapped to the specific requirements of IEC 62304. This includes document control, record keeping, training, and change control.
- Establish a risk management system — Implement a risk management system and align it with ISO 14971. The latter is a complementary standard to IEC 62304 that outlines how you identify, evaluate, control, and monitor risks during the SaMD development process.
- Develop the software development plan — Outline the scope of the SaMD project, the selected lifecycle model, roles, responsibilities, tools, technologies, and the specific IEC 62304 processes to be followed. Make sure to include information on your risk management.
Phase 2: Software requirements analysis
The phases of the SaMD-compliant development process link up with the usual SDLC, except they make software developers emphasize safety, risk management, and documentation.
Hence:
- Specify software requirements — Your development team must elicit, analyze, and document user needs, system requirements, and software requirements, including both functional and non-functional ones.
- Lay down safety requirements — Following the preliminary risk analysis, your tech partner must identify safety-related requirements to mitigate the uncovered risks. Keep in mind that each time the general requirements change, safety requirements must follow suit.
Phase 3: System design preparation
The stage of software architectural design is universal to all kinds of software development processes. However, an IEC 62304-compliant SDLC is focused on safety-first design choices that can mitigate identified risks.
- Develop software architectural design — When defining the high-level structure of the SaMD, your solution architects must factor in the safety, performance, security, and maintainability of the design.
- Document and justify your system design choices — When detailing individual software units and their interactions, your development team must look at each software unit through the prism of security, going through every potential scenario when these low-level elements could potentially impact the safety of the SaMD.
Phase 4: Software unit implementation and verification
When all the groundwork is done, your health care development team builds the software units based on the low-level design specifications and coding standards outlined in your QMS. Core to process compliance is detailed documentation of verification activities that will showcase the safety and risk controls of the implemented units.
Phase 5: Software integration and integration testing
Unit testing — the process of checking how different software components work together — is an important step for any development process. When development is guided by IEC 62304, developers verify that integrated units do not usher in new risks or undermine existing risk controls, plus document the testing activities and results.
Phase 6: Software system testing
Along with testing the software system, the development team performs end-to-end safety testing to check if the risk controls in place work as intended in the context of the entire medical device system. Like before, all testing activities and results are jotted down.
It’s impossible to eliminate all possible risks, hence there’s a concept known as residual risk. However, this remaining risk must be thoroughly evaluated to make sure it’s at an acceptable level according to predefined criteria and regulatory requirements.
Phase 7: Software Release
Besides making finishing touches to the software build, your development team prepares release documentation, including release notes and risk summary, to provide a comprehensive view of the software version, its intended use, known issues, and safety profile to both internal stakeholders and regulatory agencies.
Based on your QMS procedures, you must also get a formal authorization or sign-off from various internal stakeholders who have a vested interest in the release. Such stakeholders include your quality assurance team, development leadership, product managers, and other team members.
Phase 8: Post-market surveillance and monitoring
When your SaMD has been released, you need to establish and implement post-market surveillance (PMS) activities that are focused on both proactive and reactive measures. By proactive measures, we mean performance monitoring, security monitoring, and other activities integrated with the risk management process.
Reactive data collection from various sources, such as user reports, complaints, and adverse events, helps identify new risks and reassess existing ones. As for the standard post-release maintenance, SaMD companies must make sure it follows the same rigor as the initial development process.
How to make changes to SaMD post-market?
Software is inherently dynamic. So the SaMD is likely to undergo some changes throughout its product life cycle. Unlike conventional software updates, SaMD changes bear a higher risk profile as a single change can compromise the safety or effectiveness of the standalone software.
General principles for post-market changes
The regulatory landscape for post-market changes is still evolving and can vary by country. Yet, there are general principles for post-market changes to SaMD that stay true to all jurisdictions:
- Risk-based approach — The potential risk of a change defines the level of regulatory scrutiny.
- Impact assessment — To evaluate the potential impact of a change on SaMD safety, manufacturers must have a robust assessment process in place that considers factors such as intended use, user interface, data processing, cybersecurity, and other relevant factors.
- Change control management — Manufacturers need a well-defined change control process to systematically manage any modifications to their SaMD throughout its lifecycle.
- Post-market surveillance (PMS) — User feedback, AE reports, and other collected data inform the change, help evaluate its impact, and also demonstrate that the benefits outweigh the associated risks.
- Predetermined change control plans (PCCPs) — Manufacturers can submit a PCCP to regulatory bodies as part of the initial marketing application to proactively communicate and gain pre-authorization for specific, planned modifications to their SaMD. Examples of such pre-authorized changes can include algorithm changes for AI SaMD, performance enhancements, and security updates.
Modifying SaMD for the U.S. market
According to the FDA guidelines, the regulatory burden of the update depends on the significance of the change and may include:
- No submission required — Minor changes can be documented internally (letter to file) if they don’t have associated safety or effectiveness risk and don’t significantly modify the intended use of SaMD.
- Special 510 (k) — Some types of modifications made to SaMD with an existing 510(k) clearance are authorized through the special 510 (k) pathway.
- New 510 (k) — If the modification can have a significant impact on the safety or effectiveness of the SaMD or fundamentally alters its intended use, the manufacturer will likely be required to submit a new Premarket Notification (510(k)) before marketing the modified device.
- PMA supplement — Premarket approval supplements are commonly used to greenlight changes to existing high-risk devices approved through the Premarket Approval (PMA) pathway. The type of supplement (e.g., Panel-Track, 180-day, Real-Time, 30-day notice) hinges on the nature and significance of the change.
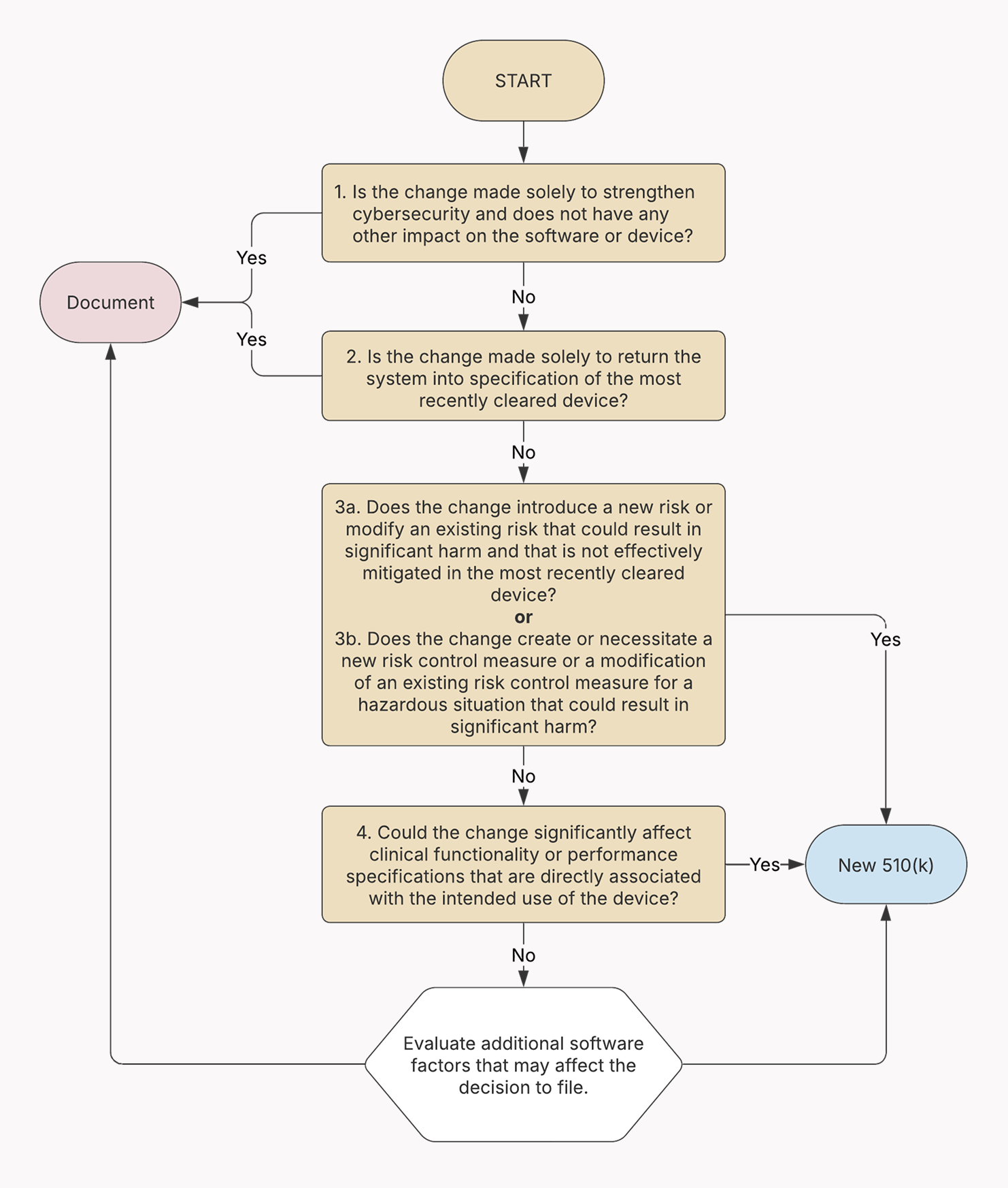
There are far more nuances to the modification process within the FDA framework. That’s why we strongly recommend reviewing the official FDA guidelines on how to decide when your modification to the SaMD requires a 510(k) submission and when it doesn’t.
Modifying SaMD for the EU market
The algorithm of SaMD modification within the MDR regulation is somewhat similar to that of the US, as all significant changes must be run by the Notified Bodies first. According to Annex X of the MDR, if the change somehow alters the SaMD’s conformity with the product's general safety and performance requirements (GSPRs) or the conditions prescribed for its use, the manufacturer must seek approval from the Notified Body that originally issued the EU type-examination certificate.
If the change is approved by the Notified Body, the manufacturer shall be notified, and the change will be formally documented through a supplement to the existing EU type-examination report.
Cybersecurity for SaMD
SaMD runs on general-purpose hardware and connects to hospital systems, cloud services, and other devices, exposing it to the same cybersecurity threats as any networked software. A breach doesn't just compromise data; it can disrupt clinical functions with direct implications for patient safety.
What the FDA requires
The PATCH Act made cybersecurity mandatory for any new device submission. Your submission must include a cybersecurity plan that outlines how you'll monitor, identify, and address vulnerabilities throughout the product lifecycle, as well as a Software Bill of Materials (SBOM) that provides a full inventory of every third-party software component in your SaMD. When a vulnerability surfaces in a widely used library, you can immediately check whether your product is affected and act before it reaches patients.
What the EU requires
Under MDR Annex I, your SaMD must meet general safety and performance requirements, including cybersecurity requirements. MDCG 2019-16 provides the detailed implementation guidance on threat modeling, penetration testing, and ongoing vulnerability management. Security is built in from the start, not added at the end.
The newer obligation to plan for is the EU Cyber Resilience Act (CRA), Regulation 2024/2847. The CRA applies to all products with digital elements sold in the EU, which includes most SaMD. It adds mandatory vulnerability disclosure to ENISA within 24 hours of an actively exploited vulnerability, and SBOM requirements that mirror what the FDA already demands.
📌 If you're planning EU market entry, you're now navigating three overlapping cybersecurity frameworks: MDR Annex I, MDCG 2019-16, and the CRA. Design your security architecture with all three in mind from the outset. Retrofitting compliance to a shipped product is significantly harder and costlier.
The regulatory approach to artificial intelligence and machine learning in SaMD
AI/ML-enabled SaMD is the fastest-growing segment of the medical device industry. The FDA has now authorized over 1,451 AI/ML-enabled medical devices, with 295 cleared in 2025 alone, up from 253 in 2024. Approximately 62% of 2025 clearances were SaMD, with radiology accounting for roughly 76% of all authorizations.
The FDA’s approach to AI-ML SaMD
The US FDA regulates AI/ML-based software intended for medical purposes the same way it regulates medical devices, even if that’s a consumer-facing app. As we know from earlier, the FDA’s classification relies on a risk-based approach to all medical devices, including AI/ML SaMD.
But unlike other SaMD, AI-powered ones are reviewed by the FDA through the lens of a total product lifecycle (TPLC) approach, which is a "cradle-to-grave" approach that demands from manufacturers ongoing oversight and evaluation of the product from the initial concept and design. From a practical standpoint, it means that:
- AI/ML SaMD developers must adhere to Good Machine Learning Practices (GMLP);
- AI/ML SaMD developers provide a detailed description of the SaMD and the algorithm it uses in the premarket submission;
- AI/ML-based SaMD manufacturers must evaluate the risk to patients of modifications to their product;
- AI/ML-based SaMD manufacturers are strongly encouraged to implement real-world performance monitoring to inform regulatory decisions.
In January 2021, the FDA published The Artificial Intelligence/Machine Learning-Based Software as a Medical Device (AI/ML SaMD) Action Plan, where they offered a more practical and in-depth look into the TPLC approach. As part of the action plan, the FDA also published the guidance on predetermined change control plans (PCCPs) within a marketing submission for AI/ML-enabled SaMD.
The MDR’s approach to AI/ML SaMD
The EU employs a dual regulatory approach to AI/ML SaMD, combining the MDR with the horizontal AI Act to cover the maximum number of AI systems. In short, the power of regulations is divided as follows:
- The MDR takes into account the associated patient risk of the AI/ML SaMD. Diagnostic or therapeutic AI/ML SaMD are typically assigned higher risk classes, such as IIa, IIb, or III, due to their potential impact on patient health.
- The AI Act further groups AI SaMD into four risk tiers, with most software falling into the high-risk category. Each risk tier has its specific obligations for conformity assessment, quality management, technical documentation, data governance, and other aspects of AI development.
Conclusion
The regulatory landscape for the SaMD technology is anything but straightforward. Various compliance pathways across regions, each with their own approaches and obligations, leave SaMD companies grappling with hurdles when planning for a successful market release. This complexity is further dialled up by the integration of AI into the standalone software.
As a digital health development company, Orangesofts helps SaMD manufacturers place their products on the market through compliant, ISO-driven discovery and development activities. We’ve enabled 300 products to find a faster route to market release and comply with the FDA, the MDR, HIPAA, GDPR, and other regulatory requirements. Contact us to find out how our SaMD development expertise can accelerate your market entry.