

For SaMD startups, IEC 62304 is not just a box-ticking exercise; it’s often a legal necessity for market approval in the US, EU, and UK. As a functional safety standard, IEC 62304 breaks down requirements for each phase of the software development lifecycle. Skip it, and you risk regulatory roadblocks, lukewarm response from investors, and, in some cases, legal fallout.
But how do you apply IEC 62304 if you’re developing software for medical devices? Find out in this blog post.
What is IEC 62304?
IEC 62304, also known as Medical device software – Software life-cycle processes, is an international standard that defines the life cycle requirements, including core processes, activities, and tasks, for developing safe and effective software for medical devices. The standard applies both to standalone SaMD and embedded software.
The standard does not cover manufacturing or hardware (that falls under ISO 13485) or general IT infrastructure, unless it’s part of the medical device.
Is IEC 62304 mandatory?
Although it’s not explicitly mandatory, IEC 62304 is a de facto standard for demonstrating compliance with software lifecycle requirements in the European Union. Not meeting IEC 62304 requirements would make it very difficult, if not impossible, for a SaMD developer to substantiate compliance with general safety and performance requirements (GSPRs) and get the proof of compliance for CE Marking.
As for the FDA, IEC 62304 is not a legally binding requirement. However, IEC 62304 compliance is strongly encouraged by the FDA and is considered a best practice framework for developing medical device software. IEC 62304 is also recognized as a consensus standard in the FDA’s 2023 Final Guidance.
IEC 62304 vs ISO 13485 vs ISO 14971
IEC 62304, alongside ISO 13485 and ISO 14971, lays out core requirements for health software development companies. Together, they form a mutually supportive framework that guides companies toward developing medical products with the highest safety standards at the core.
However, each of these guidelines is focused on a different aspect of the development process.
| Standard | Main focus | Applies to | Key goal |
|---|---|---|---|
| IEC 62304 | Software development lifecycle | Software in or as a medical device | Safe, consistent software development |
| ISO 13485 | Quality management system (traceability according to IEC 62304 is audited under Design Controls) | The entire device company or process | Quality and compliance across the organization |
| ISO 14971 | Risk management (drives IEC 62304 safety classification) | Device and software risks | Risk identification, assessment, and mitigation |
Although one is not legally required to follow these guidelines word for word, the coordinated use of the core principles of IEC 62304, ISO 13485, and ISO 14971 will help medical device manufacturers obtain regulatory approval for their products more easily and quickly.
How to implement IEC 62304 in medical device development
Let’s walk you through each step of putting IEC 62304 into action, starting with the classification.
Step 1: Classify your medical software according to ISO 14971
The standard IEC 62304 categorizes medical devices into three safety classes (A, B, and C), which define the minimum content required for a software file, including required documentation, verification, and validation activities throughout the software lifecycle.
This classification is based on the potential harm that a software failure could cause to a patient:
- Class A — If the software malfunctions, it will not cause any injury or damage to health. An example of a Class A medical device would be a scheduling module integrated into a hospital’s MRI system.
- Class B — A software failure could result in a non-serious injury. A pulse oximetry mobile app is a good example of such software. If it displays incorrect oxygen saturation levels, clinicians may hold off on immediate interventions or treatments.
- Class C — A malfunction or breakdown of the software could lead to fatal or life-threatening injuries. For example, a malfunction of an insulin pump control software might cause severe hypoglycemia or hyperglycemia, which makes it a safety-critical system.
However, IEC 62304 doesn’t provide any guidelines on how to accurately assess or quantify software-related risks. That’s when you need ISO 14971. The latter provides a formal framework for identifying, assessing, and managing risks associated with medical devices, including software. In simple words, you can then map these risks to IEC 62304’s classes.
Step 2: Define your software development life cycle (SDLC)
Onсe you’ve figured out which category your medical device falls into, you need to set up a compliant SDLC that meets the documentation rigor and process obligations of the category.
Clause 5 of IEC 62304 defines the core processes and activities for an SDLC of medical device software.
📌Keep in mind that the standard doesn’t require you to implement any specific software engineering process. You don’t need to default to a traditional approach like Waterfall. As long as you stick to the activities and requirements described in IEC 62304, you can choose Waterfall, Scrum, or any other methodology.
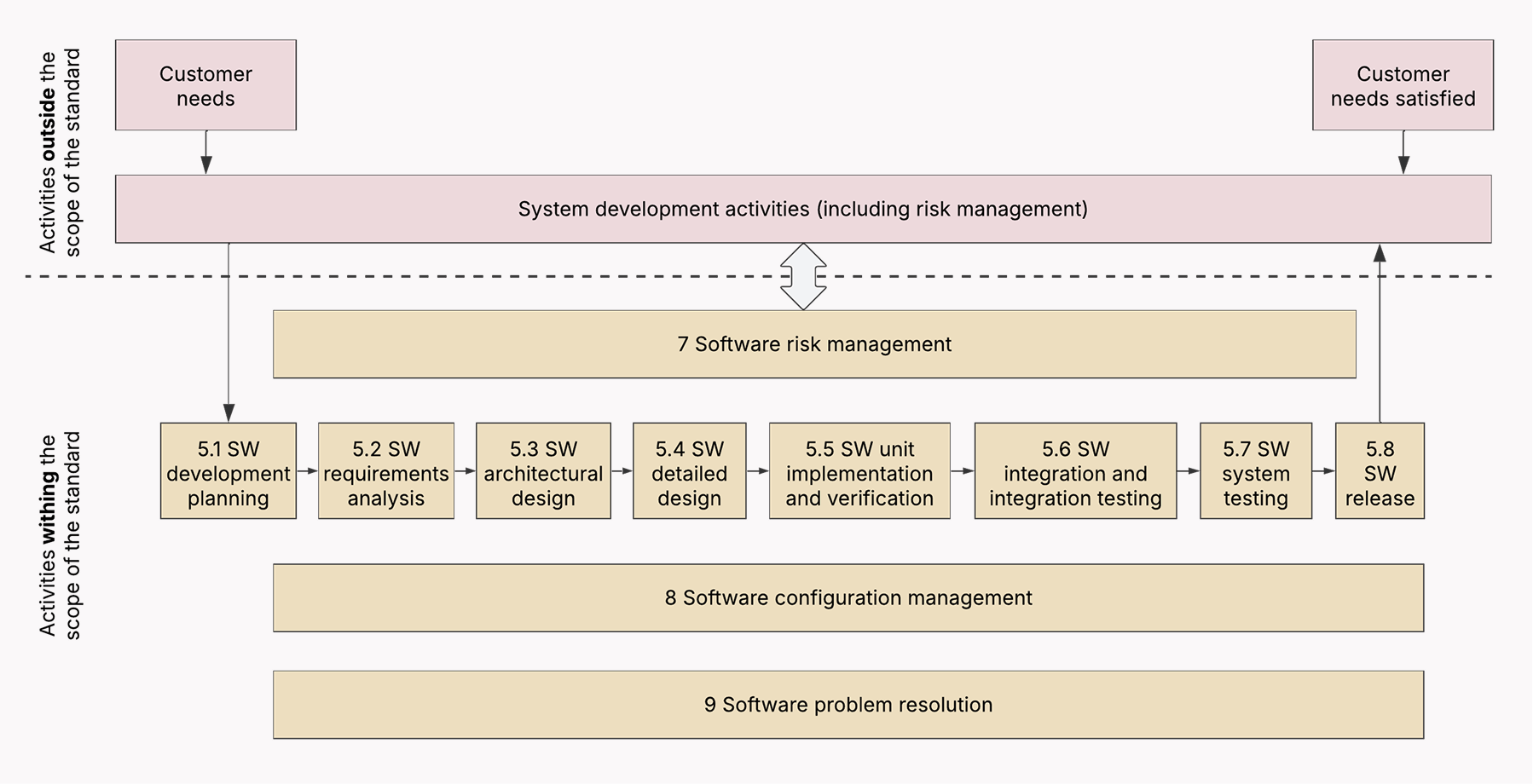
Here’s the breakdown of what activities and processes an IEC 62304-compliant SDLC must cover.
Software development planning
The standard requires software development teams to initiate the development process with a documented, version-controlled plan. The plan includes key elements such as roles, responsibilities, deliverables, a traceability strategy, and risk management.
Deliverables: Software Development Plan (SDP), configuration management plan, problem resolution plan, supplier agreement (for outsourced projects).
Software requirements analysis
Next, the team must document what the software does, including functional and safety-critical requirements. The key is to keep those requirements clear, traceable, and testable.
Deliverables: Software Requirements Specification (SRS), traceability matrix, use case or user story documentation (for Agile teams), risk management file (ISO 14971 integration).
Software architectural design and software detailed design
For a team adopting the standard, this phase means they need to design modular components, isolate high-risk components like AI in diagnostics, and thoroughly document all the interfaces. The standard expects both high-level and detailed software architecture.
Deliverables: Software Design Specification (SDS), risk control measures, data flow diagrams, detailed design documents, API documentation, algorithm descriptions (for AI/ML-based devices).
📌If your medical device software incorporates third-party or pre-made software components, you need a well-documented selection and evaluation process to prove the components don’t bring in unexpected risks or go against regulatory requirements.
Software unit implementation and verification
Under the standard, each implemented module or unit is covered with tests to ensure it meets the pre-set requirements. Class B and C devices also require proof of verification activities. For Agile teams, this stage usually includes unit tests in CI pipelines, test-driven development, and traceability in requirements.
Deliverables: Source code (with annotations/comments), unit test reports, code review records (for Class B and C).
Software integration and testing
The development team tests the interfaces between components and makes sure the integrated components work together as intended. Integration testing is documented, and the test results are checked against the architecture and requirements.
Deliverables: Integration test plan and reports, defect logs, and automated test scripts.
Software system testing
The team tests the entire software against the predefined requirements and documents the outcomes. Keep in mind that the traceability between system tests and the initial requirements is not optional.
Deliverables: System test report, traceability evidence.
Software release
Before the release, the team must double-check that all verification and validation activities are completed, issues are fixed and documented, and the software aligns with the original requirements.
Deliverables: Release notes, deployment records, final traceability matrix.
📌 IEC 62304 scales the documentation requirements based on device safety class. It means that not every deliverable listed is universally mandatory. However, in practice, even Class A devices can benefit from more detailed documentation (e.g., unit tests or design docs) to provide auditors with evidence of structured development.
Here’s how the software safety classification impacts the required development process documentation for each class.
| Software documentation | Class A | Class B | Class C | |
|---|---|---|---|---|
| Clause | Subclauses | |||
| Software development plan | 5.1.1, 5.1.2, 5.1.3, 5.1.6, 5.1.7, 5.1.8, 5.1.9 | X | X | X |
| 5.1.5, 5.1.10, 5.1.11 | X | X | ||
| 5.1.4 | X | |||
| Software requirements specification | 5.2.1, 5.2.2, 5.2.4, 5.2.5, 5.2.6 | X | X | X |
| 5.2.3 | X | X | ||
| Software architecture | 5.3.1, 5.3.2, 5.3.3, 5.3.4, 5.3.6 | X | X | |
| 5.3.5 | X | |||
| Software detailed design | 5.4.1 | X | X | |
| 5.4.2, 5.4.3, 5.4.4 | X | |||
| Software unit implementation and verification | 5.5.1 | X | X | X |
| 5.5.2, 5.5.3, 5.5.5 | X | X | ||
| 5.5.4 | X | |||
| Software integration and integration testing | All requirements | X | X | |
| Software system testing | All requirements | X | X | X |
| Software release | 5.8.4 | X | X | X |
| 5.8.1, 5.8.2, 5.8.3, 5.8.5, 5.8.6, 5.8.7, 5.8.8 | X | X | ||
| Software maintenance process | 6.1, 6.2.1, 6.2.2, 6.2.4, 6.2.5 | X | X | X |
| 6.2.3 | X | X | ||
| Software risk management process | 7.1, 7.2, 7.3, 7.4.2, 7.4.3 | X | X | |
| 7.4.1 | X | X | X | |
| X – required | ||||
Step 3: Prepare for post-market surveillance and maintenance
Regulatory bodies need proof that teams won’t let the software grow stale after it’s out the door. That’s why IEC 62304 recommends companies to double down on post-market oversight, encouraging medical device software development teams to keep a close watch on post-release field performance, updates, and issues.
According to the standard, post-market activities include:
- Problem resolution procedures — Companies need a documented problem resolution process for identifying, logging, analyzing, and resolving software anomalies. For Class B and C devices, teams have to document each step of the process.
- Change management — When making changes to the software, teams must keep to a formal workflow that defines the way those changes are assessed, approved, implemented, and verified.
- Configuration management — After deployment, the team must keep track of every version and change made to the software. Versioning, change history, and in-between traceability form a comprehensive audit trail for regulatory reviews.
- Ongoing software risk management process — Bug reports from users, adverse events, and other issues must be formally assessed according to ISO 14971.
- Field monitoring — Additionally, teams are expected to proactively track the software's behavior in real-world environments through user feedback, incident reports, usage analytics, and other methods.
📌 Teams must record post-market processes within the quality management system (QMS) and integrate them with ISO 13485 and ISO 14971 procedures.
Step 4: Establish audit and continuous improvement workflows
Compliance with the IEC 62304 standard isn’t a one-off — it’s an ongoing commitment that requires companies to regularly size up their software development and software maintenance practices.
This includes:
- Internal audits — Medical device software companies schedule regular internal audits to inspect whether their processes continue to run in conformity with IEC 62304 and with related standards like ISO 13485.
- External audits — From time to time, companies can also bring in independent auditors to get an objective evaluation of their processes.
- Feedback loops and improvements — Before running audits, companies need to set up mechanisms for collecting feedback from audits, incident reports, and team reviews. This input can then inform SOP updates, tweaks in risk management practices, and quality system refinements.
Why do your SaMD products need IEC 62304 in the first place?
In medical software development, shipping fast without guardrails isn’t just risky — it’s reckless. IEC 62304 gives health tech companies the structure, safety, and traceability needed to deliver software products that earn approval from both regulators and users.
Patient safety
Patient trust doesn’t stem solely from IEC 62304 compliance. However, the standard makes software safety an enforceable part of the development process, meaning that companies prioritize healthcare risk management over all other considerations.
Solid verification and validation approach, architecture safety, complete traceability, and controlled development process enable development teams to build software that minimizes risks and guards patient safety every step of the way.
Global market access
Regulators in key markets, including the US, the EU, the UK, Canada, and others, recognize IEC 62304 as the benchmark for SaMD development. Compliance with the standard gives companies access to global markets, allowing them to reuse documentation across markets and repurpose existing validation and verification evidence for regulatory submission in multiple markets.
Stakeholder and investor confidence
To investors, partners, and stakeholders, IEC 62304 compliance tells a story of the SaMD company that takes product security, safety, and sustainability seriously. It means that the company has committed to meeting the highest standards for quality, risk management, and long-term viability — the trio most valued by investors and stakeholders alike.
Additionally, a product built in accordance with IEC 62304 has a low risk profile, as its operational, regulatory, reputational, and liability risks have been proactively mitigated by the developer. In a competitive market like SaMD development, this level of assurance can easily become a competitive advantage.
Easier product refinement
An IEC 62304-compliant quality management system (QMS) provides the tool and structure for an agility-with-control approach. With each new code line, design element, and test case having a clear lineage, software developers can gauge the impact of any change in advance and iterate faster and safer.
Product interoperability is also made easier with IEC 62304 adherence. Modular, well-documented software design lets the development team plug the software into the existing healthcare IT system with little friction and a lower risk of disruption. Moreover, the risk-based approach allows companies to de-risk innovation, helping them incorporate AI, cloud, and other competitive technologies.
AI as a medical device: key challenges in IEC 62304 implementation
When a company integrates AI/ML features into a medical device software project, it enters a unique regulatory landscape filled with more questions than answers.
Lack of clear guidance for AI/ML development and integration
The IEC 62304 standard emerged long before the advent of AI, so it was designed with deterministic software in mind. Artificial intelligence models are non-deterministic, as their behavior is not easily predictable and traceable. In other words, aligning the AI’s dynamic nature with inherently rigid IEC 62304 requirements may feel like forcing a square peg into a round hole.
💡 Solution: Overlay the risk-based and outcome-driven approach of the IEC 62304 standard onto AI development, combining it with custom acceptance criteria and complementary standards like ISO 42001 for AI quality management.
Validation and verification complexity
While conventional software solutions can be covered with unit tests and checked against pre-defined outcomes, a similar approach falls short in AI projects. The output of AI models is probabilistic, so it’s hard to predict the exact way AI works in every possible scenario, especially in edge cases and rare clinical settings.
💡 Solution: Pair traditional testing techniques with AI-specific testing approaches, such as statistical validation of model performance across datasets, dedicated testing against edge cases, and others.
Risk management
Embedding AI features into medical device software introduces new risks to the system, including algorithmic bias, data drift, and others. Some of these risks emerge only after deployment, leaving the development team no opportunity for initial mitigation. Others, although identifiable early in the development, require tailored risk control strategies that are outside the scope of ISO 14971 alignment.
💡 Solution: Along with employing AI-specific risk controls, AI SaMD companies should use AI governance standards like ISO 42001 or frameworks such as the FDA’s AI/ML SaMD Action Plan to adapt their risk management strategy to AI.
Change management
According to IEC 62304, every update to the training data, model architecture, or hyperparameters triggers a full-blown impact analysis and regression testing. But unlike traditional software, updates to AI are more frequent, automated, and often opaque. So, the change management playbook that works for traditional software becomes unsustainable for AI-enabled SaMD products.
💡 Solution: Implementing a tiered change control process that differentiates between high-risk and low-risk changes can reduce the testing and documentation burden associated with AI medical device software. Teams can also leverage MLOps tools and ALM systems to automate validation workflows.
Traceability and explainability
Regulatory compliance with the IEC 62304 standard requires end-to-end traceability throughout requirements, design, implementation, and other software development activities. Because of their black-box nature, AI models, especially deep learning algorithms, don’t have the inherent transparency that allows developers to trace specific outputs back to the specific requirements.
💡 Solution: Companies can use interpretable models, such as decision trees and attention-based architectures, for critical functions. Explainability metrics and post-hoc explainability tools will also help create a clearer understanding of how AI models arrive at their outputs.
Develop IEC 62304 compliant healthcare software with Orangesoft
In high-stakes projects like SaMD, compliance with the IEC 62304 standard is not just a prerequisite for regulatory approval, but also a key to a more meaningful, safer, smarter, and easily scalable software solution. Whether you’re targeting FDA clearance, CE marking, or global scalability, Orangesoft has the expertise, processes, and tools to help you navigate every step of the IEC 62304 journey.
As your tech partner, we make sure you have all compliance components in place, including:
- Risk-driven SDLC design
- Seamless integration with ISO 13485 & ISO 14971
- Explainability frameworks and MLOps best practices
- Robust change control and monitoring, and more.
Beyond technical implementation, we also work with external compliance experts to make sure your SaMD solution meets both the letter and the spirit of the standard.