

If you’re a startup thinking about medical devices, regulatory compliance might seem daunting to you. You probably think that it’s going to cost too much time, money, and energy — which, in some cases, is true. To streamline your market entry and make the entire development process more manageable, you need a consistent Quality Management System (QMS).
The ISO 13485 standard is synonymous with a QMS and puts your product closer to meeting the FDA 21 CFR Part 820 and MDR requirements. Let’s examine this standard and how it’s actually implemented in real medical device development projects. All insights here are based on our 15+ years of hands-on experience in helping healthcare startups design, develop, and bring compliant devices to market.
What is ISO 13485?
Built on ISO 9001, ISO 13485 is an international consensus standard for quality management systems that lays out a framework for the development and manufacturing of medical devices. The ISO 13485 standard provides guidance on the processes, documentation, and controls companies need to demonstrate to regulatory authorities that their devices meet both regulatory and customer requirements.
From a project management perspective, the standard helps companies keep their development efforts in the utopia area, where the team can achieve the highest quality of the product in the shortest possible time and in the most cost-effective way.
Although this standard belongs to the medical device industry, any company can build on its principles to turn their manufacturing process into more structured and traceable flows.
How does ISO 13485 align with FDA 21 CFR Part 820 and MDR?
ISO 13485 is a harmonized standard that is designed to plug into core regulatory frameworks, including the FDA’s Quality System Regulation (21 CFR Part 820) in the U.S. and the EU’s Medical Device Regulation (MDR). In other words, if you’re following ISO 13485, you’re already developing in a way that checks most of the boxes required by these regulatory bodies, because they recognize it as the best practice.
Since February 6, 2026, ISO 13485:2016 will no longer be just a best practice for the FDA. In January 2024, the FDA officially embraced ISO 13485:2016 and updated its Quality System Regulation to incorporate ISO 13485:2016 by reference. This will soon make an ISO 13485-aligned QMS the most direct path to FDA approval for medical device manufacturers targeting the US market.
A similar logic applies to the MDR regulation. In this case, compliance with ISO 13485 provides a presumption of conformity for the EU MDR. However, unlike the U.S. regulatory body, the EU MDR’s mindset isn’t 100% aligned with ISO 13485, and the regulation layers additional MDR-specific requirements on top of the ISO 13485 standard.
Do software as a Medical Device (SaMD) and digital health platforms have to comply with ISO 13485?
Technically, in many jurisdictions, SaMD and digital health platforms are not legally required to comply with ISO 13485 itself, but they are mandated to have a QMS in place. ISO 13485 is a gold standard for a quality management system, and a globally recognized one. So, in reality, ISO 13485 is a de facto benchmark for both types of products, with the rigor depending on the associated risk level of the product.
Starting February 2, 2026, the FDA’s updated requirements for QMSR, SaMD, and digital health products will be audited against the new requirements aligned with ISO 13485. This makes ISO 13485 compliance not just the best device development practice for the US, but a law.
💡If your digital health platform is not classified as a medical device, such as basic wellness apps and health tracking, then ISO 13485 is not a regulatory requirement. However, even in this case, many companies still choose to implement a quality management system based on this standard. First, it’s a best practice for ensuring product safety. And second, the company won’t have to start over if it chooses to evolve into a regulated medical device.
Why ISO 13485 compliance is not just red tape for medical device companies
ISO 13485 can look like heavy paperwork from the outside. But in practice, it’s the structure that allows medical device companies to move quickly without cutting corners. The companies that scale fastest aren’t the ones skipping documentation — they’re the ones systematizing it.
Requirement for CE marking and FDA clearance
We’ve already mentioned it, but let us reiterate: ISO 13485 is a de facto or de jure regulatory requirement in many jurisdictions, including the US and the European Union. Investigators from the FDA or an EU Notified Body don’t just take the manufacturer’s word for it after seeing the innovative prototype. They need evidence and a demonstration of controlled, repeatable, and documented development processes — and ISO 13485 provides the receipts to prove it.
Also, the main point of organizing the R&D process around the standard is that the team won’t have to piece together data or specs at the last minute. Instead, they’ll have all documentation lined up so when the time comes for submission or certification, the team will already be prepared.
Product quality and safety
In 2024, there were 1,059 medical device recalls in the US alone, representing a four-year high. The reasons for recalls are different, including weak design controls, poor risk management, and other cracks. What’s common among all of these failures is that they could have been avoided had the company put ISO 13485-aligned processes in place.
ISO 13485 integrates risk management into every phase of development, so companies can prevent device failures early on. On top of that, ISO 13485 includes a series of artifacts, such as documents, records, and reports, each dedicated to ensuring one of the core pillars of product excellence — product safety, reliability, and traceability.
Access to global and local markets
ISO 13485 is an internationally recognized standard and a baseline in such core distribution markets as the European Union, Canada, the US, Australia, and even Japan. So, if a company lacks ISO 13485 certification, it’ll likely find itself locked out of market access in many locations..
Also, securing the certification can be a smart move for a multi-market strategy, since a company would already have a unified quality management system that’s valid across multiple jurisdictions. This eliminates the need to build separate quality structures for each target market and ultimately saves time and money.
Investor confidence
These days, investors don’t buy into hype or bold claims. They need proof that the product is viable and market-ready. And it’s the company’s design control documentation and risk management processes that prove that the company has the operational discipline to deliver consistently and get the sign-off from regulators when the time comes.
Nonchaotic product growth
The ISO 13485 quality management system helps the company agree on and document the smartest way to design, build, and scale the product. So, whenever a new release is looming, the team doesn’t have to go through the motions — they will have a universal way to carry it out. In a broader sense, ISO 13485 fosters a company-wide culture of quality that can be easily replicated when the company is no longer small and lean.
Key elements of ISO 13485 for medical device companies
ISO 13485 is essentially a master checklist for quality, with each section breaking down a key piece of the puzzle.
Quality Management System (QMS) — Clause 4
Clause 4 of ISO 13485 outlines the basic requirements for establishing and maintaining a Quality Management System.
General Requirements (Subclause 4.1)
According to Subclause 4.1 of ISO 13485, organizations are required to document their QMS and keep it updated and well-documented throughout the entire product development lifecycle, from design to post-market. In practice, SaMD and digital health companies fulfill this requirement by integrating a digital ecosystem that is specifically designed or configured for QMS‑type workflows.
This digital ecosystem hosts an Electronic Design History File (eDHR) that stores all the design and development artifacts, including Design Inputs, Design Outputs, risk management documentation, and others.
💡Keep in mind that code is also considered to be an artifact; that’s why the version control system and the requirement tracking system must be formally linked to the QMS as well. This way, each code commit is tied to a verified design output and a risk control measure.
Documentation requirements (Clause 4.2)
A QMS is not just a combination of processes and practices. To walk the talk, a company must support it with comprehensive documentation (about 28 documents) that describes every step of the development process. Clause 4.2 focuses on this aspect of quality management.
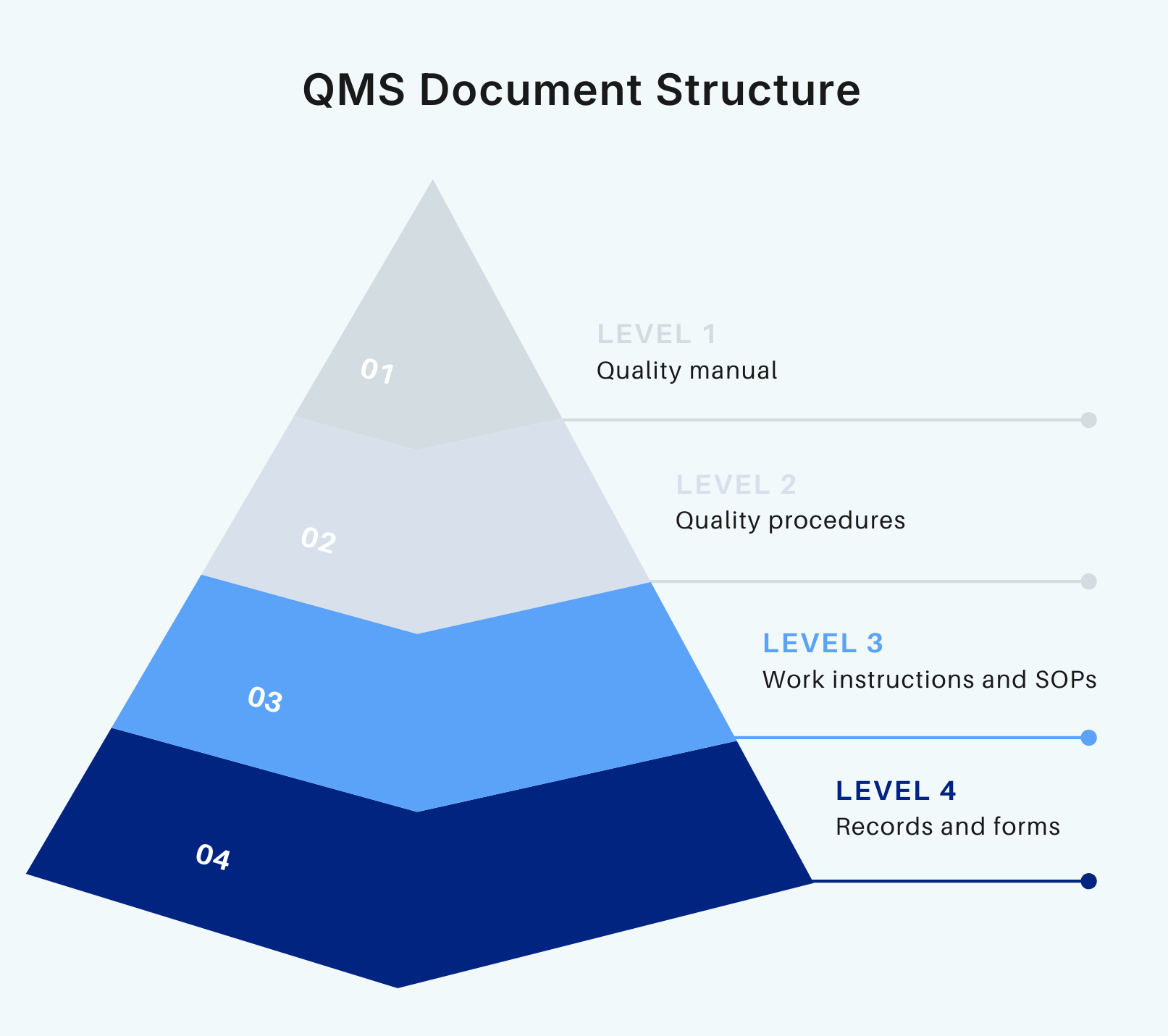
Medical Device File
According to ISO 13485, every device must have a Medical Device File or MDF to prove that it meets the regulatory and design requirements. This file typically contains such information as product descriptions, specs, product labeling, and more. What’s specific to SaMD and digital health products is that the MDF also incorporates software-related documentation, including software requirements specifications, architecture descriptions, verification evidence, and cybersecurity controls..
Quality Manual
Quality Manual is a living, central document for both internal teams and external auditors. This document explains how all processes, procedures, and roles fit into the broader QMS. Usually, the Quality Manual is the first thing auditors will study.
Document control
ISO 13485 requires that all your critical artifacts, including the Software Development Plan and Software Requirements Specifications, undergo a formal process before they can be used. The process typically includes approving documents before releasing them, maintaining version control, and implementing access control.
For SaMD and digital health companies, the same rigor applies to the code documentation. Source code, build scripts, and configuration files are just as much a part of the controlled documents as any other file.
Record control
Along with having written procedures in place, the standard requires companies to have proof that they actually followed these processes. Records, such as automated test logs, code review comments, bug reports, and deployment reports, serve as objective evidence. Records must be immutable and easily findable within a few minutes.
Management responsibility — Clause 5
Once the QMS is in place, management has to step in and implement the quality system on the ground. The core idea behind this requirement is to scale the culture of quality across the entire company, and there’s no one better than management to set the tone for it.
Key responsibilities of the executives include:
- Defining a quality policy.
- Making sure everyone in the team knows their area of responsibility.
- Setting goals and metrics tied to quality, and more.
Companies also need proof that the management followed through on commitments, so the responsibility needs to be supported by documentation.
Resource management — Clause 6
Typically, auditors check whether a company has the right people, infrastructure, and work environment to set up and maintain a safe and high-quality medical device. Under the standard, medical device companies must prove that they have:
- A team of experts to design, develop, and maintain the product (proof: employee certifications, qualifications, training records, etc.).
- Sufficient infrastructure, both physical and digital (proof: a qualified tool list).
- Controlled and secure work environment, with adequate cybersecurity and access controls.
Product realization — Clause 7
Another formal requirement of the standard, the product realization section outlines expectations for planning, designing, validating, producing, and delivering a medical device.
Let’s look at this section from a software development perspective, because it essentially describes the SDLC for medical devices:
- Planning (7.1) — Before heading into the actual development, the team must create a high-level roadmap (a Software Development Plan) that outlines stages, deliverables, review activities, and validation methods. More detailed and iterative planning happens within sprint plans.
- User needs (7.2) — The development process must be built around real-world clinical problems and user requirements, documented as a list of user needs.
- Design and development (7.3) — The process of turning user needs into a functional software product must be a disciplined and continuous cycle with built-in checks and validations.
- Purchasing (7.4) — Every piece of external technology, whether that’s AWS, GitHub, or an AI library, must be vetted.
- Production and service (7.5) — A company needs defined procedures for how it moves the builds from development to production. In tech terms, that also means that the CI/CD pipeline must be a part of the quality system, with every deployment being a traceable event.
Measurement, analysis, improvement
Quality control is not a one-off event. That’s why the standard requires companies to systematically monitor, measure, analyze, and improve their QMS and product so that they stay true to the defined requirements and needs.
Here’s a quick overview of the main data points:
- Monitoring and measurement — A medical device company tracks product performance and process effectiveness using product data (e.g., app crash reports, algorithm accuracy metrics), process data (e.g., bug report rates), and customer feedback (e.g., user support tickets, usability tests).
- Analysis of data — A medical device company builds upon the data collected to search for patterns, trends, or any early indicators of potential issues.
- Improvement through corrective and preventive action (CAPA) — The CAPA process allows the team to fix any uncovered issues and make sure they won’t reoccur.
ISO 13485 implementation process
To meet the quality system requirements, teams need to define, document, and deploy critical quality processes. Each of these processes is documented in a Standard Operating Procedure (SOP). For every standard operating procedure (or required document/process), teams repeat the following steps, approving multiple procedures either in parallel or sequentially.
Step 1: Prioritize and schedule
- Identify what processes you need at the early part of the process and when exactly you need them. Usually, document control, record control, and training are among the first procedures captured.
- For SaMD, high-priority procedures also include software development, cybersecurity, and continuous learning. It means that if a medtech company plans to collaborate with third-party vendors, it should choose those that already have structured, ISO-aligned processes in these areas.
- If the company is collaborating with the vendor for SaMD development and related services, the vendor can advise on which software, cybersecurity, and AI/ML processes are critical.
Step 2: Acquire the standard
- SaMD and digital health companies are recommended to have direct access to ISO 13485, IEC 62304, and ISO 14971. You can buy the standards on AAMI’s website.
- If the company is developing AI/ML-enabled SaMD, it also needs additional guidance, such as GMLP.
Step 3: Determine applicable clauses
- Identify the clauses that apply to your device in the quality system and product-specific sections.
- Also, make sure to address IEC 62304 software-specific clauses and ISO 14971 risk management activities that cover software hazards and AI/ML algorithms.
Step 4: Assign process owners and SMEs
- If the company handles the manufacturing process in-house, it can assign internal SMEs to monitor all SOPs and quality processes.
- If the company is collaborating with a SaMD development vendor, it can compartmentalize and have the vendor designate its internal SMEs to handle software-specific processes.
Step 5: Document processes and create SOPs
- Draft SOPs for QMS processes based on the defined regulatory requirements and device-specific needs.
- If the vendor has templates or existing SOPs for the processes they handle, make sure to integrate those SOPs into the broader framework.
Step 6: Do a gap analysis and approve SOPs
- Verify that the draft SOP meets 100% of the requirements outlined in ISO 13485, IEC 62304, ISO 14971, and GMLP guidance.
- The vendor can support the process with the evidence of compliance for the processes they perform.
- Otherwise, make sure that all SOPs, including SaMD-specific ones, qualify as controlled documents within the QMS — and approve them with e-documentation approval or document change notices.
Step 7: Train the team
- Make the approved SOPs accessible and train all team members, including new hires. Training completion must be documented in the training SOP.
- Vendors can provide training for the company’s team members who work on the vendor’s processes.
Step 8: Use SOPs and generate records
- Follow the approved SOPs as documented for each process.
- In software development terms, this step also includes the actual development of the SaMD, along with the necessary activities, such as risk management, change control, and testing.
- After each process, document all outputs as auditable records to provide proof for auditors.
Step 9: Run internal audits
- Once the company has enough records, it needs to perform a full, quality system audit internally and document the results. Usually, companies engage an outside consultant to audit the QMS before the certification.
Step 10: Prepare for certification audit and do the 1st management review
- If any nonconformities are identified during the internal audit, the company must take corrective action (CAPA) and document the objective evidence of the CAPA.
- Have the top management evaluate the effectiveness of the Quality Management System (QMS).
- Once all this is done, gather SOPs, records, and traceability matrices for the external Stage 1 and Stage 2 audits.
Step 11: Continuous improvement
- Keep improving the QMS alongside your product. For that, analyze the audit reports, monitor how the marketed medical devices perform, and collect customer feedback.
- If you uncover any gaps, make sure to update the SOPs, risk management files, and traceability matrices.
- Give extra attention to the real-world performance of AL/ML models, since this component can drift off with time, as real-world data changes.
Common challenges in ISO 13485 implementation for medical device companies
Based on our experience of working with medical device organizations, here are some common gritty challenges related to keeping up with the standard.
Adapting QMS for agile software development
At first glance, it may seem that agile and formality can’t exist in a single sentence. Auditors will look for formal design stages with clear inputs and outputs, while agile is more about continuous iteration and incremental reviews. That’s why agile adopters often struggle to prove that the sprints follow a controlled design process.
Complying with ISO 13485 doesn’t mean that a company should steer clear of agile. It means that the team needs to build a bridge between agile and regulatory compliance, which can be achieved by:
- Mapping agile to stage-gates — Adopt major milestones, like "Feature Complete" or "Release Ready", as key decision points to collect objective evidence.
- Go with a phased agile approach — This is a middle ground between agile and waterfall that allows you to run agile, but within separate phases that have clear quality gates and deliverables.
- Use dedicated tools to automate traceability — Add specialized tools that can automatically link user needs to code commits and test results. That will create a real-time audit trail without manual overhead.
Keeping documentation lean, but exhaustive for audits
“Quality documentation is killing innovation in MedTech” is a common complaint among companies. Indeed, teams often struggle to strike a healthy balance between creating just enough proof and not getting snowed under the documentation. For a team, keeping so many artifacts may feel like a full-time job that diverts their focus.
While you can’t escape meticulous record-keeping in ISO-aligned SDLC, there are ways to reduce the team's burden:
- Treat code artifacts as documentation — A well-commented pull request counts as the objective evidence required by the standard, allowing the team to replace manual documentation with automated dev records integrated directly into the normal workflow.
- Automate evidence generation — Along the same lines, a CI/CD pipeline that can automatically generate and store key records makes testing documentation, which, again, relieves the team of manual evidence production.
- Treat documents as living artifacts that are updated incrementally — Instead of rushing to update a massive doc in one go before the audit, it’s more efficient to update the risk management file/software development plan right during the sprint.
- Templatize everything — Standardize as much documentation as you can, especially when it comes to design inputs, test protocols, and risk assessments.
Aligning with other standards, such as IEC 62304 and ISO 14971
For medical device companies, ISO 13485 is not the only standard they must integrate into the development process. IEC 62304, which defines the software lifecycle requirements, and ISO 14971, a risk management guidance, are also considered must-to-adopt for medtech projects, as they add technical depth to a QMS.
With so many requirements on their hands, companies often get lost in the standards and fail to approach this trio of standards as complementary parts of a unified compliance ecosystem. This often results in duplicated efforts, such as when the company documents separate risk logs and design records for each standard.
To weave ISO 13485, IEC 62304, and ISO 14971 into a single narrative, we recommend that medtech companies:
- Go with a unified framework, not parallel processes — Where possible, don’t split procedures into separate activities (e.g., risk management procedure), and integrate software development activities from IEC 62304 into the design and development process under ISO 13485.
- Adopt a single, centralized risk management file — When the team logs all risk management activities into a single file, it’s easier to gather all software-related risks, hardware hazards, and system-level issues into one to provide a traceable story for auditors.
- Use tools that support cross-standard traceability — Specialized QMS platforms and application lifecycle management (ALM) tools can centralize requirements, design outputs, risk controls, test cases, and verification results across ISO 13485, IEC 62304, and ISO 14971.
Integrating AI/ML-enabled SaMD into a quality management system
Initially, a QMS is designed for static medical device manufacturing, with predictable inputs and outputs as well as one-time validation. AI and ML models continuously learn and adapt based on real-world data after deployment, which runs counter to the traditional validation and verification approaches covered by ISO 13485.
Here’s how companies can make use of innovative technologies and simultaneously demonstrate compliance with applicable regulatory requirements:
- Integrate Good Machine Learning Practices (GMLP) into the SDLC — Doing this allows companies to account for unique, AI-specific challenges in safety, performance, and regulatory compliance, as well as add more structure to AI development processes.
- Make sure your change control process is advanced — Introduce a pre-approved, automated change control protocol into the QMS, since the traditional one is too reactive for adaptive algorithms.
- Opt for algorithms that support the XAI approach — Avoid using models with black-box logic, as the logic of such models can’t be explained and documented to the extent required by the standard.
How Orangesoft supports ISO 13485-compliant development
As a healthcare tech partner, our team works with medtech companies across the US and Europe, taking over their both regulated and non-regulated digital initiatives.
Our proven track record of SaMD projects includes:
- A medication management app that achieved a 41% boost in medication adherence among polypharmacy users (under NDA).
- An FDA-compliant remote monitoring platform that scaled to 50K+ users.
- An FDA-ready connected care platform that cut infrastructure costs by 30%.
We also developed medical imaging platforms, compliant early-stage digital health products (HIPAA, FDA, GDPR, MDR), clinical workflow automation tools, clinical trial matching platforms, AI assistants for chronic patients, digital platforms for post-acute recovery, and other solutions.
For the majority of our projects, we help companies design a fit-for-purpose QMS aligned with ISO 13485, IEC 62304, ISO 14971, and the FDA’s new Quality Management System Regulation (QMSR). Overall, we are comfortable developing according to our internal SDLC SOPs, aligned with regulatory standards, and integrating into the client-specific SOPs.
Here’s how we support the ISO 13495 international standard and compliance in practice:
- Evidence of compliance for the processes we perform and end-to-end documentation support, including all required SOPs, templates, test reports, risk files, and other documentation necessary for regulatory purposes.
- Integration of ISO 14971 and GMLP into the SDLC, ensuring risk tracking, model versioning, and explainability for AI/ML components.
- Formative testing to spot usability or safety issues early and mitigate risks.
- Full verification and validation support to make sure all functional and performance tests are traceable and auditable.
- Support with audit preparation, including ISO, FDA, and MDR audits.
- Post-market monitoring support, including AI/ML model drift monitoring, real-world performance tracking, and continuous improvement.
It isn't red tape, it's the structure
When it comes to ISO 13485, we prefer to think of it not as a limitation or a documentation requirement. We associate the standard with the guardrails that keep the product (and patients) safe. Ultimately, ISO 13485 isn’t just about compliance. It allows the medtech company to build a company-wide culture of quality and to build confidence with regulators and investors, setting the stage for the product’s long-term success in the market.